|

|
Main /
RESEARCH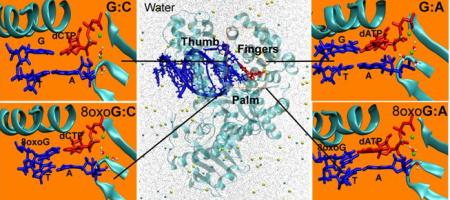 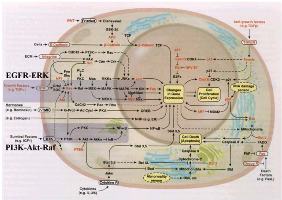 HOME | MEMBERS | RESEARCH SUPPORT | PUBLICATIONS | MEDIA | RESEARCH | MOBILE RESEARCH PROJECTS A. Molecular Carcinogenesis: In search of a scale for genotoxicity Summary: High accuracy synthesis in DNA replication and repair is a prerequisite for transmitting genetic information reliably from generation to generation. This venerable surveillance job of accurate duplication despite occasional DNA damage falls in large part on DNA polymerases which, during DNA replication and repair, incorporate nucleotides to the primer strand complementary to the template bases. Since mutations stemming from polymerase errors can result in permanent genomic change that may lead to human diseases like colon, skin, or lung cancer and premature aging, understanding fidelity mechanisms is of great scientific and biomedical importance. The fidelity of DNA polymerases refers to their ability to choose correct nucleotides from a pool of structurally similar molecules. Across the many known polymerases, fidelity varies from one to nearly one-million base errors per one-million nucleotides synthesized. Our prior studies on polymerase beta suggested an orchestration of molecular events essential to the nucleotide incorporation process. We plan to extend these studies to other polymerases, incorporation of mismatches, and carcinogens. Our modeling platform is expected to establish a new paradigm to view molecular carcinogenesis and establish a scale for genotoxicity of carcinogens. Such information will help better understand fidelity mechanisms, a fundamental problem of great importance, including the processes by which damaged DNA at the active site affects the incorporation and extension of mispairs that can eventually lead to mutations and human diseases. Our studies have immediate applications to the diagnostics, and eventually treatment via polymerase inhibitors, of human diseases caused by defective repair of DNA, like various cancers and premature aging. B. Cancer Therapeutics: Drug resistance in receptor tyrosine kinase inhibitors Background: Cancer therapy directed at specific, frequently occurring molecular alterations in signaling pathways of cancer cells has been validated through the clinical development and regulatory approval of agents such as Herceptin for the treatment of advanced breast cancer and Gleevec for chronic myelogenous leukemia and gastrointestinal stromal tumors. Tumor cells have been known to over express tyrosine kinases to have a survival advantage. Therefore, supplementing a tyrosine kinase inhibitor in conjunction with chemotherapy or radiation can enhance the effectiveness of the treatment. In recent years, several pharmaceutical companies have developed small-molecule inhibitors of the epidermal growth factor receptor's (EGFR's) tyrosine kinase activity. Several agents are currently in clinical development, differing in their specificity for EGFR and other members of the HER2 family, their potency, and the reversibility of their interaction with the ATP binding site in the receptor tyrosine kinase domain of the receptor. Among the EGFR inhibitors in clinical development, Iressa (ZD1839, Astra Zeneca) has progressed furthest, and its development is influencing that of other targeted therapeutics. The detailed knowledge of the structural mechanism underlying activation and inhibition of receptor tyrosine kinases by small molecule modulators can help address the issue of drug resistance that often depreciates the long-term effectiveness of receptor tyrosine kinase inhibitors [2]. Three broad mechanisms may result in the restoration of kinase activity: (1) decreased intracellular levels of the drug; (2) increased expression of the kinase; or (3) intrinsic changes in the kinase that affect its drug interaction or kinase activity via point mutations in the receptor tyrosine kinases may impair drug binding at the active-site. Mechanism (3) has perhaps been most widely studied, and based on clinical studies, appears to be the most common mechanism of resistance. Crystal structures of receptor tyrosine kinases in the presence or absence of ATP analogues and inhibitors have suggested at the atomic level, how the spectrum of point mutations can induce drug resistance. However details of the mechanisms involved remain unknown, and quantitative models to describe the drug resistance phenomenon do not exist. Project Scope: The objective of the proposed project is to elucidate the molecular basis for drug resistance caused by single nucleotide polymorphism effects on the receptor tyrosine kinase domain of the epidermal growth factor receptor by employing computational methods to perform structural analysis, molecular dynamics simulations, free energy computations, electronic structure calculations, and mixed quantum/classical simulations. Broader Impact: Detailed and quantitative mapping of the polymorphisms on the structure of the drug binding domain and kinase signaling activity can reveal why agonists or antagonists become inactive against the mutated receptor. The theoretical and computational approach adopted here helps catapult structure-based drug design approaches to new heights by enabling the formulation of molecular mechanisms for drug resistance to cancer therapy, and opens the door to the rational design of improved anti-cancer drugs, customized for each patient. C. Targeted drug delivery: Integrated Multiscale Modeling Summary: Because the therapeutic effects of many drugs are accompanied by serious toxicity and severe side effects, new experimental methods of targeted drug delivery are under development. One strategic approach is to couple drug packaging inside sustained-release microreservoir liposome or polymer drug carriers with ligand-receptor mediated binding of the drug carrier vehicle to the vascular endothelium. Drug carrier binding can be increased selectively by incorporating into the vehicle, membrane specific molecules that adhere to receptors or ligands that are uniquely expressed or overexpressed within diseased tissue relative to normal tissue. Thousands of drug molecules to treat a disease can be packaged within each microcarrier vehicle. The selective delivery of drugs with varying potencies, drug loading, drug retention, rate of drug release, and dose-related toxicity can be modulated by careful design and manufacture of the specific carrier vehicle for a particular drug to increase the dose of the drug reaching the diseased tissue while simultaneously decreasing the dose reaching normal tissue. Data currently available from state-of-the-art experiments indicate that microcarrier drug delivery has the potential to add a valuable asset to the pharmacological armamentarium for clinical therapeutics. The interplay between the motion, binding and transport dynamics that ultimately define targeted drug delivery is highly complex and not easily discerned from experiments due to the macroscopic and microscopic nature of the critical events occurring at multiple length and time scales. Development of an integrated multiscale model of targeted microcarrier drug delivery will improve our understanding the physicochemical, hydrodynamic, and binding interactions that determine drug transport and delivery. We propose to develop a multiscale hydrodynamic transport model of transvascular drug delivery out of a microcarrier "encapsulated spherical droplet" or "bead" initially delivered into the vasculature. The microcarrier is deformable and laden with a diffusible drug (e.g., antineoplastic, antiarrythmic) or gene therapy agent and its surface is also coated with ligands specific for receptors preferentially expressed on the luminal surface of endothelium within the specific tissue of interest. We address the following specific issues: (1) drug loss from the microcarrier vehicle from the point of injection in the bloodstream to the point of binding to the target tissue; (2) that lateral diffusion of receptors and ligands and its effect on cell membrane dynamics; (3) drug transport from the microcarrier to the target cell. Our models will incorporate the time-dependent change in concentration of the diffusible species out of the microcarrier and into the target tissue. The modeling will account for motion of the microcarrier within sufficient proximity of the endothelial surface for binding to occur, and for the continual transport of the diffusible agent to the free stream of blood before and after microcarrier adhesion as well as transport across the contact surfaces of the microcarrier and the endothelium. Because of the interdependency of the coupled transport events occurring between the flowing blood and the endothelial surface, the modeling will incorporate and bridge multiple scales (e.g., continuum mechanics, molecular mechanics and molecular dynamics, stochastic methods accounting for microcarrier arrest versus rolling). For the macroscopic modeling of momentum and mass transfer, a front tracking scheme with adaptive mesh refinement will be employed. To unravel the microscopic pathway and associated energetics of the transport mechanism at atomic resolution, we propose to employ a combination of long-time molecular dynamics simulations and novel Monte Carlo based sampling methodologies. A series of experiments to assess transport of representative diffusible drug species through elastic microporous lipid bilayers with varying degrees of transmural pressure applied to induce stretch will accompany the modeling for validation purposes. This novel work will serve two major purposes. First, it will provide important results for application to intelligent design of drug delivery systems for the targeted treatment of diseases where no integrative modeling of the multiple micro- and macro-scale mechanics currently exists. Second, it will provide important new computational methods for modeling of biofluid dynamics and biotransport systems integrating multiple length and time scales. D. Signal Transduction in Biochemical Networks After Genomics, What? It was realized that simply knowing the sequence wasn't going to answer many of the biological questions. There has been a growing effort to figure out in a concerted way, underlying mechanisms of biological processes, by theoretical, computational, and experimental means, and to ask questions that allow us to get to a level of understanding where prediction, control, and design is feasible. We are involved in developing and applying multiscale algorithms that enable predictive modeling of complex system behavior across multiple scales encompassing enzyme catalysis, protein conformational changes, single molecule manipulations, and signal transduction. There is imminent necessity for such physically-based modeling ranging from electronic, molecular to microscale because there is a large gap to be bridged between genomics/ bioinformatics approaches and predictions of subcellular response. Such modeling will also be impactful in bridging branches of experimental biology, namely structural biology with single molecule studies, biochemistry, and macroscopic kinetics, and establish a self-consistent set of tools for studying signal transduction in biological systems. A focus group that I lead and mentor is the SysB (systems biology) group. The group consists primarily of undergraduate students from Penn; recruitment is also through the The Greater Philadelphia Bioinformatics Association (GPBA), and Penn's NSF AMP program. Current focus of the SysB group is to explore signaling network at the subcellular level, their dynamic behavior, robustness, sensitivity to mutations in individual enzymes etc. The applications are largely synergestic with the rest of our research program. For example, in one application, we are applying network analysis of EGFR signaling pathways to transcend our predictions at the molecular level into the larger context of subcellular response. In another application, we are exploring the link between calcium release in response to receptor binding and an internalization process known as endocytosis. This latter project has overlap with our drug delivery as well as our cancer therapeutics initiatives. We are developing new algorithms to integrate molecular dynamics with quantum mechanical methods, hydrodynamics (Navier Stokes equations and the equivalent in membranes known as Ginzburg-Landau dynamics) with Monte carlo and applying these to our signaling networks. We are also developing robustness analysis tools based on principal component analysis. Students interested in being part of the SysB group need to have a solid mathematics and physical sciences background in addition to knowing basic cell biology. Familiarity with computing is a plus. Thanks to Penn's CETS, our software is installed in the SEAS public domain so that any person with a seas email account can run our simulations. Students of the SysB group work out of the numerous Linux machines maintained by CETS. The examples explored by the SysB group are featured in BE459. The independent study (BE490) is also open to interested students. |