|

|
Main /
BE459559FinalProjectMiniSymposiumDate: Friday 05/08/2009
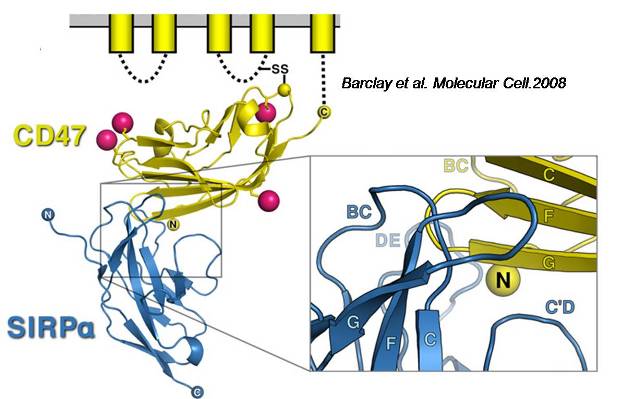 Abstract: Automated docking method helps to predict the bound conformations of flexible ligands to macromolecular targets. We decided to use this method to understand a specific mechanism of the immune system in human macrophages. Integrin-associated protein (IAP) or CD47, is ubiquitously expressed in all tissues and serves as a ligand for signal regulatory protein (SIRPá), an immune inhibitory receptor found on macrophages. CD47-SIRPá signaling results in the inhibition of phagocytosis of normal hematopoietic cells by macrophages. Applications of this self-inhibition of phagocytic engulfment for drug delivery will necessitate studying isolated CD47 and SIRPá proteins. Hence, an understanding of the x-ray crystallography structure and molecular dynamic docking simulations can help us predict the interaction of CD47 (45-60 kDa), with SIRPá (90-120 kDa), and we will also gather insight with the calculation of free energy change upon binding. SIRPá belongs to a class of a membrane protein family called "paired receptors" that comprise of several genes coding for proteins with similar extracellular regions but radically different transmembrane/cytoplasmic regions with different (activating or inhibitory) signaling potentials. Whereas SIRPâ with a 90% identity has a short cytoplasmic region and associates with a transmembrane adapter protein DAP12 gives an activating signal for phagocytosis. Therefore, we decided to test DAP12 as a ligand for SIRPá to observe the difference in the energy that an activator of phagocytosis would produce and compare that to an inhibitor (CD47).
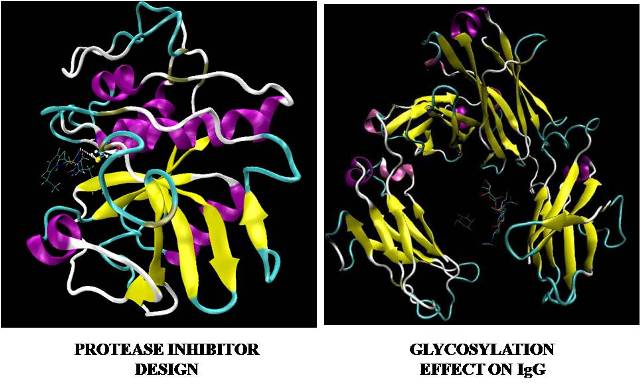 Abstract: Alongside with crystal structure, molecular modeling through docking software serves as a computational tool to understand drug action in an atomistic level. Two examples are presented to illustrate its applicability in chemical drug and biopharmaceutical R&D. A. Protease inhibitor design Human Cathepsin L has been implicated in the viral entry process in the infection pathway of several viruses such as SARS and Ebola. Cathepsin L-like proteases are found in many parasites and have been shown to play vital role in diseases including malaria, leishmaniasis and amoebiasis. Thus development of inhibitors of human cathepsin L may provide a gateway into finding therapeutic treatments against these illnesses. Recently a novel inhibitor with thiocarbazate scaffold has been identified from a high throughput screening campaign in Penn Centre for Molecular Discovery. Molecular docking is performed to understand the interaction between cathepsin-L and thiocarbazate to lay down a platform for rational drug design. B. Glycosylation effect on IgG The consistent production of therapeutic antibody with a human glycoform profile has been a challenge to the biopharmaceutical industry. Experimental data from Clinical through Phase III production batches of a monoclonal antibody shows a shift in the G0F, G1F, G2F and SA1 species in the IgG-Fc regions. These differences in IgG-Fc glycosylation may result in immunogenicity. Therefore, a relevant human IgG-Fc glyoform will be used to understand differences in IgG-Fc glycosylation. Structural configurations and binding energies of the beta-D-mannose ligand to the crystal structure of the human IgG-Fc fragment glycoform of MNF2 will help understand shifts in glycoform species.
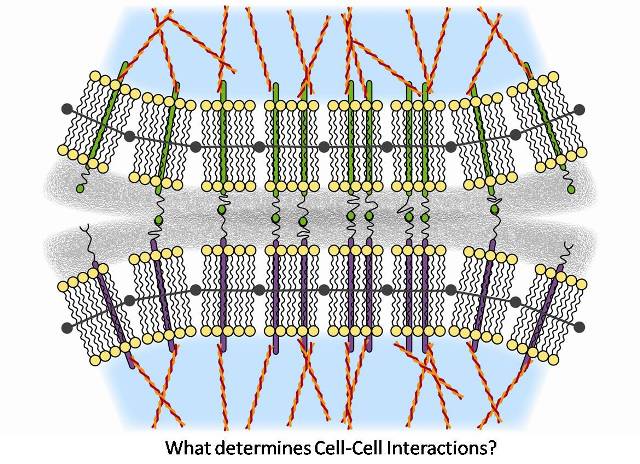 Abstract: This project approaches the issue of cell adhesion from two standpoints: an established thermodynamic model, and Monte Carlo simulation. The thermodynamic model, first introduced by Bell in 1984, examined the equilibrium state of two potentially adherent cells by minimizing the Gibbs free energy of binding given constraints on area of contact, cell separation distance, and the number of bound receptors. Cells were assumed to adhere by interactions between separate receptor populations on each cell surface. Free and bound receptor chemical potentials and steric hindrance from the glycocalyx of each cell were incorporated in the minimization. The receptor-receptor bond was modeled as a spring, and this approximation was added to the bound receptor chemical potential. The degree of cell adherence was eventually characterized by the three constraining parameters. This model is compared to repeated simulations, based on Metropolis Monte Carlo, of two cells with separate receptor populations that undergo binding and dissociation to an equilibrium state. Steric repulsion is incorporated by introducing an energy penalty due to compression of the glycocalyx. The Monte Carlo model will also explicitly consider the shape change of the membrane allowing the model to investigate the effects of membrane stiffness. The comparison of these two techniques will provide valuable insight into the validity of thermodynamic models and Monte Carlo simulations in various adhesion regimes, and expose interesting points for experimental exploration.
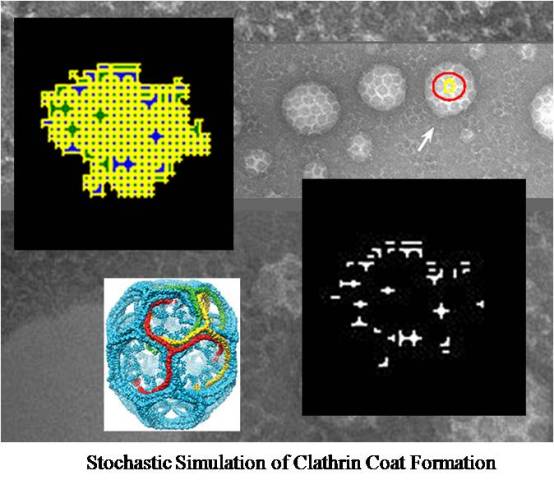 Abstract: This paper attempts to investigate the thermodynamic conditions under which Clathrin can form its unique three-dimensional structure and to describe the dynamics of Clathrin assembly using computer simulation and stochastic modeling. Clathrin, a ubiquitous protein in eukaryotic cells, plays a central role in receptor-mediated endocytosis as well as the stabilization of spindle fibers during mitosis. Its three-pronged structure enables the protein to form planar sheets and curved baskets which can become the scaffolding for intracellular vesicles. After a vesicle buds into the cytoplasm the coat rapidly disassembles allowing the Clathrin to recycle while the cargo within gets selected for a variety of fates. The author’s simulate the Clathrin lattice by implementing a modification of the cellular Potts model using the software CompuCell 3D. Specifically, because the molecule’s conformation to membrane invagination requires that defects form in its ordinary planar organization, CompuCell is used to explicitly and dynamically quantify lattice defects and characterize their formation. Data is collected using a range of thermodynamic parameters and conclusions are drawn on the importance of defect formation in Clathrin dynamics. This work presents potential application to the understanding and manipulation of RME signaling. In particular, cancer cells show an inability to attenuate growth signals via the regulation of surface receptors, which could be due to a break down in clathrin-mediated endocytosis.
 Abstract: Imatinib is an orally adminstered drug used to treat a number of types of cancer, including chronic myelogenous leukemia (CML) and gastroinstestinal stromal tumors (GISTs). After receiving FDA approval in May 2001, it was marketed under the name Gleevec and crowned by TIME magazine as the “magic bullet” of cancer treatments. Gleevec functions primarily as a tyrosine kinase inhibitor, selectively targeting abl proteins of cancer cells to prevent tumor cell proliferation. While this therapy has been highly effective, often limiting or halting disease progression, drug resistance continues to be a problem in a minority of patients. The nature of imatinib resistance is largely unknown, and molecular simulations may play a significant role in elucidating the underlying mechanisms. One proposed explanation by the Consortium for Bioinformatics and Computational Biology at the University of Minnesota suggests that a T315I and P-loop mutation of the bcr-abl kinase domain contributes to resistance. In this proof-of-concept study, a wildtype and a mutated bcr-abl protein were each inhibited by imatinib and visualized in AutoDockTools. Using qualitative observations of the ligand-kinase conformation and quantitative parameters such as dock score, the plausibility of this resistance mechanism was evaluated. A literature review was also conducted to determine the role of molecular simulations in the current state of the field and new breakthroughs in the design of imatinib and other cancer drugs.
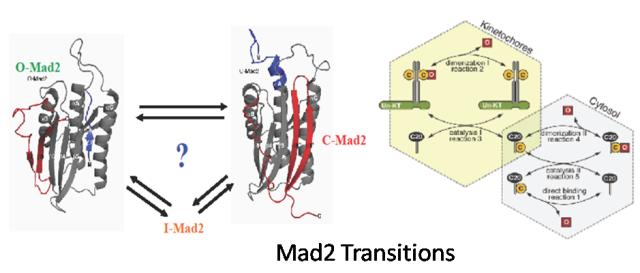 Abstract: Mad2 is the diffusible effector of the Spindle Assembly Checkpoint (SAC) which ensures faithful chromatin separation in mitosis. It is metamorphic, having 2 stable forms with a shared core and rearranged termini. One form binds to its effectors much more rapidly. Mad2 binds its upstream and downstream effector molecules in a mode reminiscent of a seatbelt. In order either to achieve this binding or to interconvert from one state to the other, it is presumed to partially unfold. We will perform NAMD simulations with the Langevin thermostat on. We will run simulations to investigate the conformational stability of each form at high and low temperatures, hoping to discover clues of the intermediate conformation(s) between them. We will use also a set of equations to study systematically the Mad2 binding under different conditions, and compare the predictions with experimental results.
 Abstract: HER3/ErbB3 is a member of the EGF/ErbB family of receptor tyrosine kinases that is essential for initiating signal transduction and mediation of processes involving cellular development, homeostasis and differentiation. HER3 is the only member of the ErbB family that is classified as a pseudokinase as it is thought to lack tyrosine kinase activity due to amino acid substitutions in the conserved kinase domain. Here we construct homology models of the HER3 kinase domain using MODELLER and investigate its active site bound to ATP and Mg2+ for use in simulations to understand the molecular basis for the catalytic uniqueness of HER3 and to determine the differences between HER3 and the other ErbBs at the atomistic level. HER3 derived from EGFR, HER2, HER4, and combined template of EGFR and HER4 showed dominant clusters of conformations, favorable Discrete Optimized Protein Energy (DOPE), and good stereochemical properties. The top four structures from the different templates were all very similar. Proximal residue analysis of active site showed both conservations and differences in residues. In particular, H740 of HER3’s αC helix differed, explaining HER3’s inability to attain the active conformation. These results produce quality structures for HER3 kinase and rationalize its unique kinase activity.
|