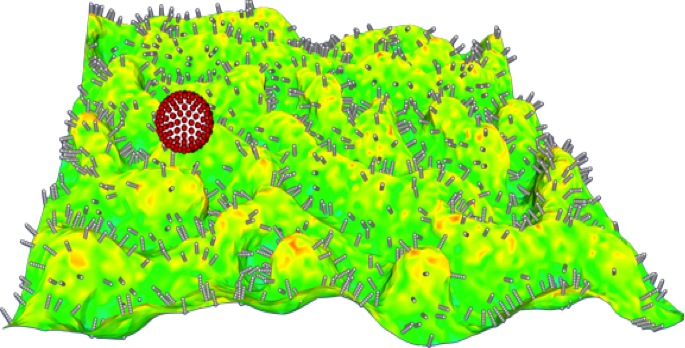
AI-Enabled Multiscale Modeling
Quantitative Systems Biology; Quantitative Systems Medicine; Quantitative Systems Pharmacology
Our research interests lie at the interface of biophysics and cellular engineering. Our goal is to provide quantitative mechanistic characterization of complex biological and physiological systems and formulate quantitatively accurate microscopic models for predicting the interactions of various therapeutic agents with innate biochemical signaling mechanisms, while retaining molecular specificity. We employ several computational algorithms ranging from techniques to treat electronic structure, molecular dynamics, Monte Carlo simulations, stochastic kinetic equations, continuum field methods, and complex systems analyses in conjunction with the theoretical formalisms of statistical and quantum mechanics, hydrodynamics, and applied machine learning and high performance computing in massively parallel architectures.
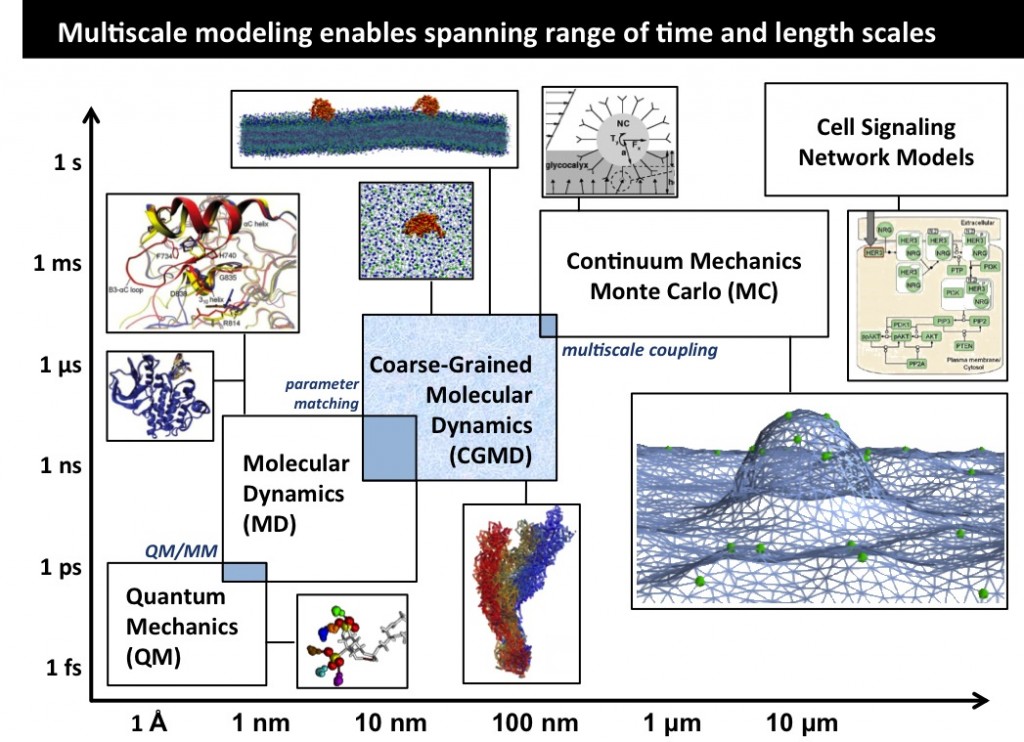
Multiscale Modeling of Intra-Cellular Trafficking
Cell Membranes are know to remodel in a wide variety of biological processes including endocytosis and cell motility. Our research probes different length scales of membrane biophysics by utilizing a suite of computational models from quantum and atomistic models up to large mesoscale models of membrane elasticity. Certain cell membrane proteins are known to both sense and induce curvature, this curvature generation may play a major role in their function and localization in vivo. Atomistic modeling of protein-lipid interaction provides a mechanistic molecular-level picture of how proteins can induce curvature at the nanoscale.
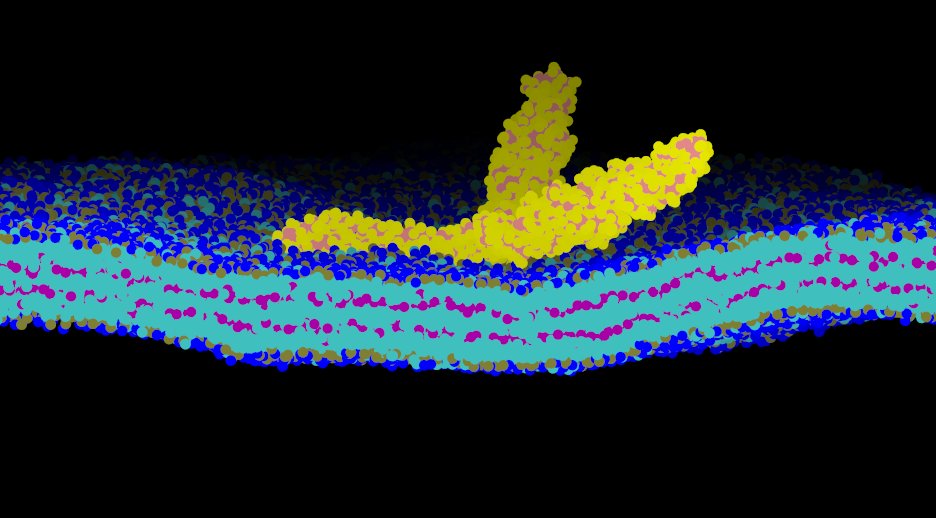
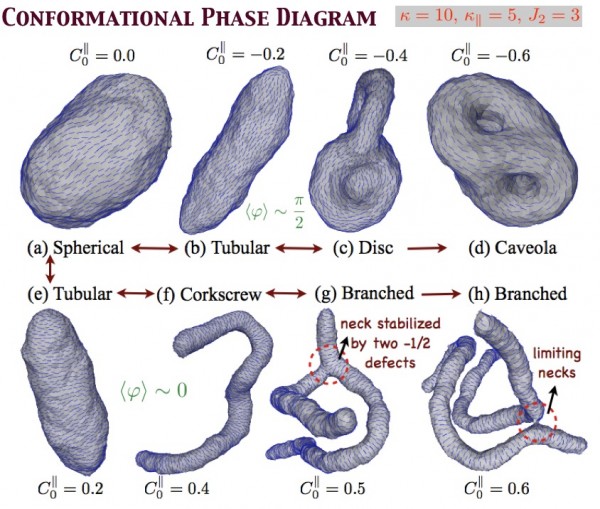
In order to translate the effects of protein-membrane interaction at the nanoscale to emergent cellular scale phenomena such as tubulation or vesiculation of cell membranes, we employ continuum field based stochastic models of protein-membrane interactions.
Mathematical Modeling in Insilico Oncology
We develop multiscale computational modeling and simulation approaches in our laboratory combining models in structural biology and those in systems biology. The overall aim is to build quantitative models of signaling networks while retaining the crucial elements of molecular specificity, which appear to be highly relevant to profiling and predictive modeling of clinical cancer mutations in many transformed cell lines. We utilize current and emerging experimental and computational methods, and applied machine learning, particularly focusing on hybrid and multiscale methods, which we have developed, and highlight several applications in cell signaling with quantitative and predictive capabilities. The scope of such models range from delineating protein-protein interactions to describing clinical implications in the context of mutations in various cancer types
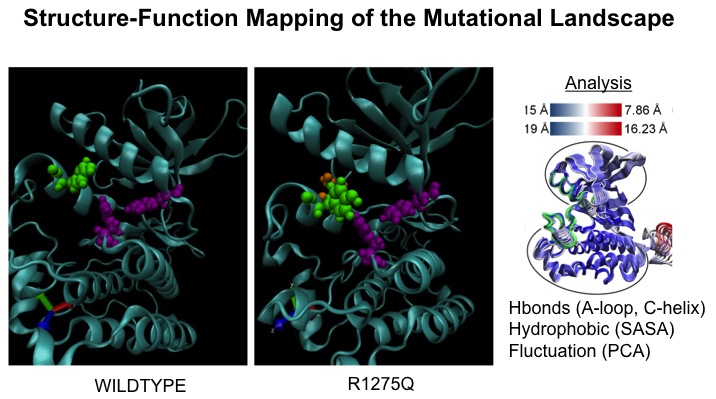
Understanding Mutation-driven Activation of Oncogenic Proteins in Cancer Patients: Activating mutations in the kinase domain of anaplastic lymphoma kinase (ALK) have been implicated in the childhood cancer, neuroblastoma. Using molecular dynamics-based methods, we investigate how these mutations promote constitutive kinase activation. We analyze changes in solvent-accessible surface area, hydrogen-bond occupancies, and water density fluctuations, compare principal components of motion, perform extensive free energy perturbation studies, and apply scoring functions and seauence and structure based features including those from protein language models to characterize each mutation's mode of activation. Each mutation is classified as activating by hydrophilic destabilization of the αC-helix, activating by hydrophilic destabilization of the A-loop, activating by disruption of the “hydrophobic patch”, or not activating. We compare our results to in vitro kinase assays measuring kinase activity for each mutation and to proliferation assays which determine which activating mutations are also driving cellular proliferation. We suggest that a combination of statistical data-driven modeling and computational modeling may be useful a useful tool in predicting the activating potential of under-studies oncogenic proteins.
Next Generation Pharmacodynamic Models for Targeted Drug Delivery
Dynamics for Functionalized Nanocarrier Adhesion to Cell Surfaces in the Presence of Hydrodynamic Interactions: Vascular delivery of antibody-functionalized nanocarriers (50 nm-1 mm in size) to selectively bind targeted surface receptors expressed by endothelial cells is a viable therapeutic strategy in systems pharmacology. However, optimal nanocarrier design involves the appropriate selection of their size, shape, and antibody density on their surface. Additionally, the choice of appropriate tethers/linkers bridging the antibodies to the nanocarrier surface depends on the physiological microenvironment, including hemodynamics and rheological properties of blood flow in the microvasculature as well as the state of the endothelial cells (which governs expression of targeted receptors such as intracellular adhesion molecule-1, ICAM-1). A model-based optimization of this design can be used to guide the achievement of targeting selectivity and specificity in vivo. We address the important aspects of nanocarrier adhesive dynamics in terms of how they impact binding selectivity and specificity. We employ techniques ranging from direct numerical simulations for fluctuating hydrodynamics, brownian dynamics, and generalized Langevin dynamics. We emply AI-enabled model integration including physics informed neural networks to efficiently solve for physiologically based pharmacokinetic models at multiple scales.
Physiologically Validated Models for Adhesion of Functionalized Nanocarriers to Endothelium in Targeted Drug Delivery: Targeting nanocarriers (NCs) loaded with drugs and probes to precise locations in the body may improve the treatment and detection of many diseases. Generally, to achieve targeting, the surface of the NC is coated with affinity ligands to enhance binding to molecules present on the cells of interest. Optimization of ligand density is a critical parameter in controlling NC binding to target cells. Other tunable properties include NC size and shape, surface chemistry (e.g., tethers of various lengths and stiffnesses) and deformability (such as gel-based NCs). Our objective is to develop a computational platform for modeling the targeting of functionalized NCs to optimize experimental design protocols for drug delivery. Recently we introduced a computational methodology based on Monte Carlo and the weighted histogram analysis method to calculate the absolute binding free energy between functionalized NC and endothelial cell surfaces. The calculated NC binding free energy landscapes have been validated against measurements performed in vivo as well as in cell-culture experiments. The effect of antibody surface coverage of NC on binding has been studied using the model that accounts for physiological factors such as hydrodynamic forces (e.g., shear stress) and resistance due to glycocalyx layers. We validated the simulations against three distinct classes of experiments: in vivo targeting in mice, targeting in cultured cells under flow, and binding assays using Atomic Force Microscopy. While higher ligand density always leads to higher binding avidity, it is not always the most effective. In a recent study, we investigated how NC avidity affects targeting to the pulmonary vasculature comparing NC binding to quiescent endothelium and inflamed endothelium. Expectedly, NC avidity was controlled by ligand density, with the higher avidity NCs demonstrating greater pulmonary uptake than lower avidity NCs in both naive and pathological mice. However, in comparison with high-avidity NCs, low-avidity NCs exhibited several-fold higher selectivity of targeting to pathological endothelium. This finding was translated into a Positron Emission Tomography imaging platform that was more effective in detecting pulmonary vascular inflammation using low-avidity NCs. Our computational modeling revealed that elevated expression of ICAM-1 on the endothelium is critical for multivalent anchoring of NCs with low avidity, while high-avidity NCs anchor effectively to both quiescent and activated endothelium. These results provide a new paradigm whereby computational model predictions are used to optimize NC targeting in pathological vasculature. In current work on targeted drug delivery, we are developing multiscale computational strategies to incorporate NC adhesion to live cells, where membrane compliance and fluidity play dominant roles. In particular, we investigate the explicit effects of membrane stiffness and deformation on the NC adhesion landscape.